
Click here to see all images
April, 2019
Case of the Month
Clinical History: A 46-year-old asymptomatic female, former smoker, with a recent diagnosis of breast carcinoma in-situ was found on chest computed tomography scan to have a 1.2 cm right middle lobe rounded solitary nodule. PET scan reported a right middle lobe nodule, mildly hypermetabolic, possibly neoplastic, without other evidence of malignancy or metastatic disease. The patient was directed to thoracic surgery and resection was performed.
Quiz:
Q1. Which of the following is true about this lesion?
- The patients are often asymptomatic
- It occurs more frequently in women
- It is a very common endobronchial lesion
- It frequently involves chest wall and pleura
Q2. Which chromosomal rearrangement is diagnostic of this lesion?
- t12;16 FUS-DDIT3
- 8q PLAG1 rearrangement
- 2p ALK rearrangement
- t12;22 EWSR1-CREB1
Q3. This lesion has been given numerous names in the past, including which of the following?
- Myohyaline hamartoma
- Plasma cell granuloma.
- Indolent myxoid pulmonary tumor
- Myxovascular endobronchial tumor
Answers to Quiz
Q1. A
Q2. C
Q3. B
Q2. C
Q3. B
Diagnosis
Inflammatory myofibroblastic tumor
Discussion
Inflammatory myofibroblastic tumor (IMT) is a mesenchymal neoplasm of intermediate biological potential composed of myofibroblastic cells associated with variable inflammatory infiltrate of plasma cells, lymphocytes and eosinophils. This tumor was given numerous names in the past, notably plasma cell granuloma and inflammatory pseudotumor. IMT can occur at any anatomic site, and comprises less than 1% of all lung tumors. It usually presents as a peripheral lung lesion, but in about 10-20% of cases can be endobronchial. Approximately half of patients with IMT are symptomatic. Local symptoms may be secondary to local growth and obstruction of the airways. Systemic symptoms are variable and include fever, night sweats, weight loss, accompanied by laboratory findings of anemia, elevated erythrocyte sedimentation rate and thrombocytosis. IMT has potential for local recurrence and rarely metastases.
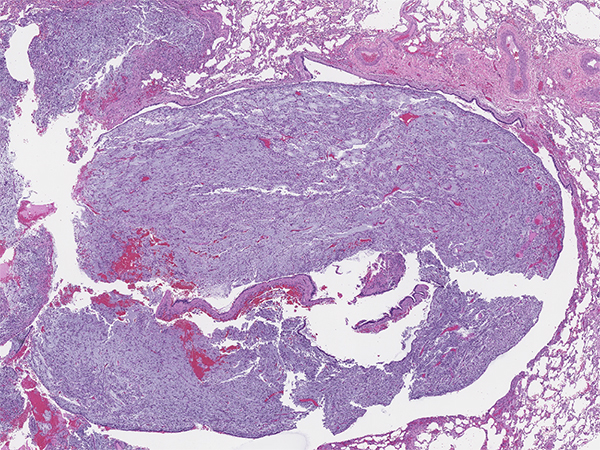
IMT is a morphologically heterogenous neoplasm (Figures 1-6). Common morphologic patterns have been reported, each eliciting a broad differential diagnosis such as; inflammatory-rich, storiform reminiscent of nodular fasciitis and sclerotic scar-like. Cellular morphology varies from spindle, stellate to large ganglion-like cells with prominent nucleoli. The immunoprofile is rather non-specific and includes variable expression of smooth muscle actin (Figure 7), muscle-specific actin and desmin. Cytokeratin expression can be detected in about 30% of cases. ALK gene rearrangement resulting in expression of a chimeric fusion protein is found in approximately 50% of cases, mostly in children and younger adults. Detection of either ALK gene rearrangement or ALK protein overexpression (Figure 8) is diagnostically helpful. In contrast, ALK-negative IMT can be diagnostically challenging and essentially represents a diagnosis of exclusion. The differential diagnosis in ALK-negative cases include a wide spectrum of reactive/inflammatory processes on one end such as sclerosing mediastinitis, IgG4-related disease, pulmonary hyalinizing granulomas, and on the other end of spectrum malignant processes such as MALT lymphomas and sarcomas.
Numerous translocations partners of ALK gene have been reported, most commonly TPM3 (t(1;2)) and TPM4 (t(2;19)). Recent studies further demonstrated tyrosine kinase abnormalities in all of thoracic IMTs. About 30% of the tumors showed abnormalities outside of ALK kinase, including gene fusions involving ROS1, RET and NTRK3. These findings may provide additional diagnostic tools and a rationale for targeted therapeutic strategies with FDA-approved tyrosine kinase inhibitors.
The prognosis of IMT is very good when completely resected, but it should be kept in mind that recurrences and distant metastasis do occur, most commonly in tumors of abdominal and retroperitoneal origins. The prognosis may be influenced by large tumor size, incomplete resection and absence of ALK rearrangement.
Take home message for trainees: Inflammatory myofibroblastic tumour may overlap morphologically with a large number of inflammatory and malignant sarcomatoid processes. The immunoprofile is non-specific, but ALK gene rearrangement and protein expression are diagnostic of IMT. .
IMT is a morphologically heterogenous neoplasm (Figures 1-6). Common morphologic patterns have been reported, each eliciting a broad differential diagnosis such as; inflammatory-rich, storiform reminiscent of nodular fasciitis and sclerotic scar-like. Cellular morphology varies from spindle, stellate to large ganglion-like cells with prominent nucleoli. The immunoprofile is rather non-specific and includes variable expression of smooth muscle actin (Figure 7), muscle-specific actin and desmin. Cytokeratin expression can be detected in about 30% of cases. ALK gene rearrangement resulting in expression of a chimeric fusion protein is found in approximately 50% of cases, mostly in children and younger adults. Detection of either ALK gene rearrangement or ALK protein overexpression (Figure 8) is diagnostically helpful. In contrast, ALK-negative IMT can be diagnostically challenging and essentially represents a diagnosis of exclusion. The differential diagnosis in ALK-negative cases include a wide spectrum of reactive/inflammatory processes on one end such as sclerosing mediastinitis, IgG4-related disease, pulmonary hyalinizing granulomas, and on the other end of spectrum malignant processes such as MALT lymphomas and sarcomas.
Numerous translocations partners of ALK gene have been reported, most commonly TPM3 (t(1;2)) and TPM4 (t(2;19)). Recent studies further demonstrated tyrosine kinase abnormalities in all of thoracic IMTs. About 30% of the tumors showed abnormalities outside of ALK kinase, including gene fusions involving ROS1, RET and NTRK3. These findings may provide additional diagnostic tools and a rationale for targeted therapeutic strategies with FDA-approved tyrosine kinase inhibitors.
The prognosis of IMT is very good when completely resected, but it should be kept in mind that recurrences and distant metastasis do occur, most commonly in tumors of abdominal and retroperitoneal origins. The prognosis may be influenced by large tumor size, incomplete resection and absence of ALK rearrangement.
Take home message for trainees: Inflammatory myofibroblastic tumour may overlap morphologically with a large number of inflammatory and malignant sarcomatoid processes. The immunoprofile is non-specific, but ALK gene rearrangement and protein expression are diagnostic of IMT. .
References
WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart, Travis & al., WHO Classification of Tumours, 4th Edition, Volume 7, 2015.
American Journal of Pathology, TPM3-ALK and TPM4-ALK Oncogenes in Inflammatory Myofibroblastic Tumors, Lawrence & al., 157(2), 377-84, 2000.
Histopathology, ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours, Yamamoto & al., 69; 72-83, 2015.
American Journal of Pathology, TPM3-ALK and TPM4-ALK Oncogenes in Inflammatory Myofibroblastic Tumors, Lawrence & al., 157(2), 377-84, 2000.
Histopathology, ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours, Yamamoto & al., 69; 72-83, 2015.
Contributors
Martin Chevrier, M.D. BSc.
Thoracic Pathology Fellow
University of Pittsburgh Medical Center, Pittsburgh, PA, USA
Sanja Dacic, M.D. PhD.
Professor of Pathology
University of Pittsburgh Medical Center, Pittsburgh, PA, USA
Thoracic Pathology Fellow
University of Pittsburgh Medical Center, Pittsburgh, PA, USA
Sanja Dacic, M.D. PhD.
Professor of Pathology
University of Pittsburgh Medical Center, Pittsburgh, PA, USA