
Click here to see all images
January, 2020
Case of the Month
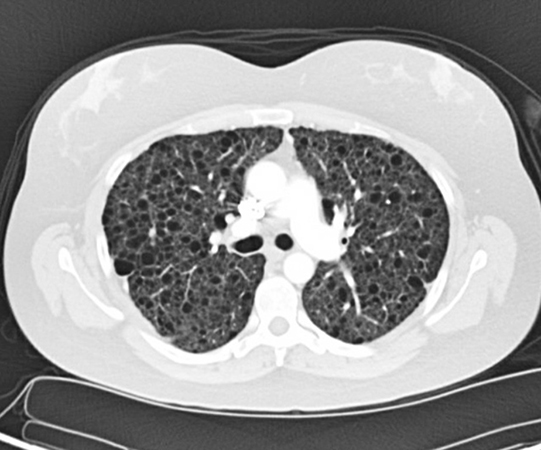
Clinical History: A 29-year-old female never-smoker with a history of a loculated left pneumothorax 2 years ago presented with a right-sided spontaneous pneumothorax prompting requirement for pigtail chest tube placement. A high-resolution computed tomography scan (HRCT) performed 3 months before the current presentation (Figure 1) showed innumerable bilateral subcentimeter thin-walled lung cysts.
Quiz:
Q1. Based on her prior HRCT findings, which description is most appropriate for her condition?
- The disease near exclusively involves women in a sporadic setting but may develop in men in a familial setting.
- It is an autosomal dominant condition also characterized by the presence of cutaneous hamartomas and kidney neoplasms.
- There does not appear to be a gender predilection in this condition, and nearly all affected individuals have a history of current or prior cigarette smoking.
- It is an idiopathic disease and could be associated with AIDS in patients with perinatally acquired HIV, hypergammaglobulinemia, Sjögren's syndrome and other autoimmune conditions.
Case continued...
The patient subsequently underwent VATS for therapeutic and diagnostic purposes, and a wedge resection of the superior segment of the right lower lobe was performed. Representative images of the wedge resection are shown in Figures 2-4.
Q2. Which immunohistochemical stain is least useful to confirm the diagnosis?
- Smooth muscle actin
- S-100
- ER/PR
- HMB-45
Q3. Which of the following clinical condition(s) is/are associated with this entity?
- Renal angiomyolipoma
- Chylothorax
- Seizures/cognitive dysfunction
- All of the above
Q4. Mutations of which gene are associated with this entity?
- FLCN
- Tuberous sclerosis complex (TSC1, TSC2)
- BHD
- BRAF
Answers to Quiz
Q1. A
Q2. B
Q3. D
Q4. B
Q2. B
Q3. D
Q4. B
Diagnosis
Lymphangioleiomyomatosis
Discussion
Lymphangioleiomyomatosis (LAM) is a rare multisystem disorder and can be either sporadic or in association with Tuberous Sclerosis Complex (TSC). TSC is an autosomal dominant genetic syndrome presenting with a constellation of symptoms including neurocutaneous lesions and multiple hamartomatous lesions in the kidney, skin, brain, retina and lung. The sporadic form of LAM almost exclusively affects women of reproductive age, and with an estimated incidence of about 3-8 cases per million women. In the TSC background, LAM (TSC-LAM) may occur in men, but it is also seen more frequently in women. Both sporadic LAM and TSC-LAM harbor mutations in the tuberous sclerosis genes (TSC2>TSC1). These genes code for hamartin and tuberin proteins which lead to the formation of an intracellular dimer that inhibits mammalian target of rapamycin (mTOR), a kinase that promotes cell growth and proliferation.
Patients with LAM usually present with progressive dyspnea due to deterioration of pulmonary function, which may be worsened by exertion, as the disease process progresses. However, acute onset of dyspnea and chest pain in the setting of cystic lung disease warrants a suspicion of spontaneous pneumothorax due to LAM as in the current case. This is especially important in female patients of childbearing age, or under a clinical context of tuberous sclerosis syndrome with an obstructive or mixed obstructive and restrictive pattern on pulmonary function evaluation. Pleural effusion, hemoptysis, pulmonary hemorrhage, chylothorax and lymphadenopathies may also occur. The main radiological manifestation of LAM is the presence of multiple (>10), thin-walled, round, well-defined air-filled lung cysts of various sizes (2 to 40 mm in diameter) distributed evenly in both lungs.
LAM belongs to the family of neoplasms with perivascular epithelioid differentiation (PEComa), and both sporadic and TSC-associated forms share macroscopic and histologic features. The typical macroscopic appearance of LAM shows lung parenchyma studded with multiple cysts distributed diffusely and uniformly in all lobes. Histopathologically, it is characterized by the proliferation of LAM cells (Figure 4) that may surround lymphatic vessels, veins, and bronchioles, and may form overt or subtle nodular cell clusters in the edge of the pulmonary cysts. The LAM cells lack cellular atypia, necrosis and mitotic activity, and are characterized by two distinctive cytomorphologic appearances; the plump spindle-shaped cells with eosinophilic cytoplasm resembling vascular smooth muscle cells located in the center of the nodular lesions, and cuboidal cells with features of perivascular epithelioid cells (PECs: clear to granular, lightly eosinophilic cytoplasm) seen in the periphery. Representative images of immunostains are shown in Figures 5-9. The LAM cells demonstrate strong cytoplasmic immunoreactivity to melanocytic markers including HMB-45 (Figure 5), melan A, Microphthalmia transcription factor (Mitf), and smooth muscle markers including α-smooth muscle actin (Figure 6) and desmin (Figure 7). The LAM cells may also show nuclear labeling to estrogen receptors (Figure 8) and progesterone receptors (Figure 9). Additionally, a histologic finding known as Multifocal Micronodular Pneumocyte Hyperplasia (MMNPH) may coexist with LAM, in particular, in patients with TSC-LAM. MMHPH is described as well-demarcated, multifocal nodules composed of a tubulopapillary proliferation of type II pneumocytes with enlarged vesicular nuclei, occasional conspicuous nucleoli lining the alveolar walls, and thickened septa. These features may mimic atypical adenomatous hyperplasia (AAH) and adenocarcinoma in situ (AIS).
While initial assessment and follow-up are crucial in the management of LAM, delayed recognition/diagnosis of the disease is common. Suspicion of LAM usually starts with the detection of multiple cysts on HRCT, but this alone is insufficient to establish a definitive diagnosis. Thus, guidelines have been established and recommend a categorical evaluation of clinical, imaging and/or histopathological findings on a surgical lung biopsy to establish a probable, possible or definite diagnosis for LAM. Sirolimus (Rapamycin) has been shown to be an effective therapy in patients with LAM, especially in those with chylous effusion and those with a decline in lung function. However, lung transplantation may be the only therapeutic option for patients with advanced LAM or those refractory to mTOR inhibitors.
Take home message for trainees: always include LAM in the differential diagnosis of bilateral lung cysts in a woman.
Patients with LAM usually present with progressive dyspnea due to deterioration of pulmonary function, which may be worsened by exertion, as the disease process progresses. However, acute onset of dyspnea and chest pain in the setting of cystic lung disease warrants a suspicion of spontaneous pneumothorax due to LAM as in the current case. This is especially important in female patients of childbearing age, or under a clinical context of tuberous sclerosis syndrome with an obstructive or mixed obstructive and restrictive pattern on pulmonary function evaluation. Pleural effusion, hemoptysis, pulmonary hemorrhage, chylothorax and lymphadenopathies may also occur. The main radiological manifestation of LAM is the presence of multiple (>10), thin-walled, round, well-defined air-filled lung cysts of various sizes (2 to 40 mm in diameter) distributed evenly in both lungs.
LAM belongs to the family of neoplasms with perivascular epithelioid differentiation (PEComa), and both sporadic and TSC-associated forms share macroscopic and histologic features. The typical macroscopic appearance of LAM shows lung parenchyma studded with multiple cysts distributed diffusely and uniformly in all lobes. Histopathologically, it is characterized by the proliferation of LAM cells (Figure 4) that may surround lymphatic vessels, veins, and bronchioles, and may form overt or subtle nodular cell clusters in the edge of the pulmonary cysts. The LAM cells lack cellular atypia, necrosis and mitotic activity, and are characterized by two distinctive cytomorphologic appearances; the plump spindle-shaped cells with eosinophilic cytoplasm resembling vascular smooth muscle cells located in the center of the nodular lesions, and cuboidal cells with features of perivascular epithelioid cells (PECs: clear to granular, lightly eosinophilic cytoplasm) seen in the periphery. Representative images of immunostains are shown in Figures 5-9. The LAM cells demonstrate strong cytoplasmic immunoreactivity to melanocytic markers including HMB-45 (Figure 5), melan A, Microphthalmia transcription factor (Mitf), and smooth muscle markers including α-smooth muscle actin (Figure 6) and desmin (Figure 7). The LAM cells may also show nuclear labeling to estrogen receptors (Figure 8) and progesterone receptors (Figure 9). Additionally, a histologic finding known as Multifocal Micronodular Pneumocyte Hyperplasia (MMNPH) may coexist with LAM, in particular, in patients with TSC-LAM. MMHPH is described as well-demarcated, multifocal nodules composed of a tubulopapillary proliferation of type II pneumocytes with enlarged vesicular nuclei, occasional conspicuous nucleoli lining the alveolar walls, and thickened septa. These features may mimic atypical adenomatous hyperplasia (AAH) and adenocarcinoma in situ (AIS).
While initial assessment and follow-up are crucial in the management of LAM, delayed recognition/diagnosis of the disease is common. Suspicion of LAM usually starts with the detection of multiple cysts on HRCT, but this alone is insufficient to establish a definitive diagnosis. Thus, guidelines have been established and recommend a categorical evaluation of clinical, imaging and/or histopathological findings on a surgical lung biopsy to establish a probable, possible or definite diagnosis for LAM. Sirolimus (Rapamycin) has been shown to be an effective therapy in patients with LAM, especially in those with chylous effusion and those with a decline in lung function. However, lung transplantation may be the only therapeutic option for patients with advanced LAM or those refractory to mTOR inhibitors.
Take home message for trainees: always include LAM in the differential diagnosis of bilateral lung cysts in a woman.
References
Abbott GF, Rosado-de-Christenson ML, Frazier AA, et al. Lymphangioleiomyomatosis: Radiologic-Pathologic Correlation. Radiographics 2005;25:803-28.
Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A 2000;97:6085-90.
Guinee D, Singh R, Azumi N, et al. Multifocal micronodular pneumocyte hyperplasia: a distinctive pulmonary manifestation of tuberous sclerosis. Mod Pathol 1995;8:902-6.
McCormack FX, Gupta N, Finlay GR, et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med 2016;194:748-61.
Ryu JH, Moss J, Beck GJ, et al. NHLBI LAM Registry Group: The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med 2006;173:105-11.
Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A 2000;97:6085-90.
Guinee D, Singh R, Azumi N, et al. Multifocal micronodular pneumocyte hyperplasia: a distinctive pulmonary manifestation of tuberous sclerosis. Mod Pathol 1995;8:902-6.
McCormack FX, Gupta N, Finlay GR, et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med 2016;194:748-61.
Ryu JH, Moss J, Beck GJ, et al. NHLBI LAM Registry Group: The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med 2006;173:105-11.
Contributor
Treah May S. Sayo, MD
Pulmonary Pathology Research Fellow
Department of Pathology
Massachusetts General Hospital, Boston, MA
Julian A. Villalba, MD
Resident in Pathology, PGY-4
Department of Pathology
Massachusetts General Hospital, Boston, MA
Mari Mino-Kenudson, MD
Professor of Pathology, Harvard Medical School
Director, Pulmonary Pathology Service
Massachusetts General Hospital, Boston, MA
Pulmonary Pathology Research Fellow
Department of Pathology
Massachusetts General Hospital, Boston, MA
Julian A. Villalba, MD
Resident in Pathology, PGY-4
Department of Pathology
Massachusetts General Hospital, Boston, MA
Mari Mino-Kenudson, MD
Professor of Pathology, Harvard Medical School
Director, Pulmonary Pathology Service
Massachusetts General Hospital, Boston, MA