
Click here to see all images
March, 2023
Case of the Month
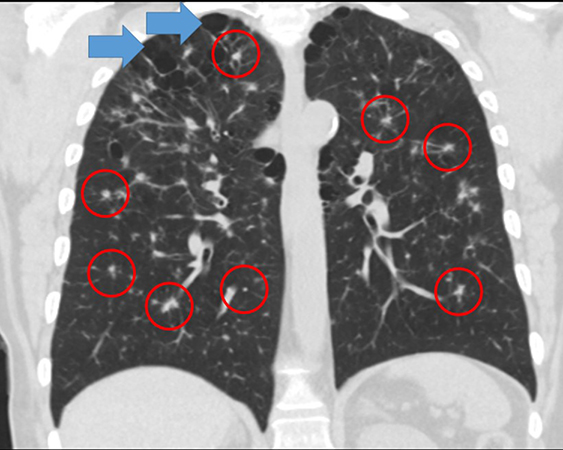
Clinical History: A woman in her 60s presented with cough. Chest CT showed moderate emphysema, reticulonodular changes bilaterally, and numerous bilateral irregular and spiculated nodular densities scattered throughout both lungs (Figure 1) significantly increased in size and number since a CT performed 4 years prior (a lung biopsy performed at that time had been non-diagnostic). There was radiologic concern for metastatic disease or an atypical infectious process. Her pulmonary function tests show mild obstruction, no restriction and mildly reduced DLCO. She was a 30-pack-year current smoker. A second CT-guided core needle biopsy was performed. Photomicrographs from the biopsy are shown in Figures 2-4 (Figures 2 and 3: H&E, Figure 4: CD1a).
Q1. What is the etiology of this disease?
- It is considered a form of idiopathic interstitial pneumonia
- Connective tissue disease
- Hypersensitivity pneuymonitis
- Smoking
Q2. What are the likely outcomes of this process?
- The nodules will incrase in size and ultimately invade the chest wall
- It will either regress, remain stable or progress to interstitial fibrosis
- If the process is not treated, distant metastases will develop to the bones and skin
- The patient will develop diabetes insipidus
Q3. Which of the following is true regarding smoking-related pulmonary Langerhans cell histiocytosis in adults?
- Most patients have evidence of clonal Langerhans cell proliferations in the skin and bones
- It is standard therapy to treat the smoking-related pulmonary form of this disease with BRAF inhibitors
- Only a minority of cases have a BRAF mutation
- Bronchoalveolar lavage (BAL) is a common and reliable means of making this diagnosis
Answers to Quiz
Q2. B
Q3. C
Diagnosis
Discussion
Pulmonary Langerhans cell histiocytosis is a smoking-related form of interstitial lung disease defined by the presence of clusters/sheets of Langerhans cells within the interstitium of the lung. Older (obsolete) terms for this disease include eosinophilic granuloma, histiocytosis X, Langerhans cell granulomatosis and eosinophilic granulomatosis. Langerhans cells (not to be confused with Langhans giant cells, found in true granulomas) are antigen-presenting dendritic cells that originate in the bone marrow, eventually enter the circulation and make their way to the skin (where they are found in the epidermis) and lung. Unlike ordinary macrophages (histiocytes), which ingest (phagocytose) and destroy antigens, the function of Langerhans cells is to detect antigens and present them on their surface to other cells of the immune system. Langerhans cells are named after Paul Langerhans (1847-1888), a German pathologist who was mentored by Virchow. When Langerhans described these cells in 1868 using a gold chloride technique, he thought they were nerves of the skin (he was wrong). Also named after Paul Langerhans are the islets of Langerhans (pancreas), Langerin (CD207), and the Langerhans cell layer (granular cell layer) of the skin. Langerhans cells are positive for the immunohistochemical markers S-100, CD1a, and Langerin (CD207).
Cigarette smoking causes the vast majority of cases of lung-limited Langerhans cell histiocytosis in adults; most patients are heavy cigarette smokers. Suri and colleagues have proposed that the lesional Langerhans cells are associated with local proliferation of the cells as opposed to another mechanism of accumulation (such as enhanced recruitment and survival, or delayed apoptosis). These authors speculate that the smoking-induced form of PLCH is a biologically distinct histiocytosis variant that is more consistent with a reactive rather than a clonal proliferative process.
The vast majority of adult smokers with pulmonary disease have involvement limited to the lung. Cases with smoking-related pulmonary Langerhans cell histiocytosis and histologically confirmed extra-pulmonary disease are very rare. Although lung involvement (sometimes very striking) can occur in Langerhans cell histiocytosis in children, the disease that occurs in children is truly neoplastic and is a completely different clinical entity than pulmonary Langerhans cell histiocytosis in adult smokers.
Pulmonary Langerhans cell histiocytosis is a disease of adult cigarette smokers. It can occur at any age and affects both sexes. Many individuals with pulmonary Langerhans cell histiocytosis are asymptomatic. In patients who undergo lung biopsy, the disease is often discovered incidentally when lung nodules are noted during a chest CT performed for another reason. The frequency of asymptomatic pulmonary Langerhans cell histiocytosis in heavy smokers is probably greatly underestimated. Pulmonary Langerhans cell histiocytosis has been reported as an incidental finding in lobectomies for lung cancer. In symptomatic patients, typical symptoms are non-productive cough (most common), dyspnea, fatigue, weight loss and fever. Patients with advanced scarring may develop cough and shortness of breath. Patients with cysts may present with a pneumothorax (pleuritic pain and acute shortness of breath). Physical examination is usually unhelpful. Crackles and clubbing are both uncommon.
The usual chest x-ray findings are diffuse bilateral reticulonodular infiltrates with normal or increased lung volumes. Chest CT findings include bilateral lung nodules that are often centrilobular, cavitary and are almost invariably tiny (millimeter-sized). They may be so numerous as to raise the possibility of disseminated mycobacterial or fungal infection or disseminated malignancy on imaging. This differential diagnosis often prompts a lung biopsy. Bilateral thin-walled cysts may also occur. These are usually - but not always - bilateral and upper and mid-lung predominant. Relative sparing of lung bases is often cited in the imaging literature. Bilateral ground-glass opacities, probably attributable to macrophages and/or smoking-related interstitial fibrosis (SRIF). Theoretically, the combination of nodules and cysts might be so characteristic that the diagnosis can be made on the basis of chest CT alone and biopsy can be avoided. In practice, although radiologists often suggest the diagnosis in classic cases, most cases are biopsied for confirmation. Pathology is the gold standard for this diagnosis.
On pathology, the diagnosis requires the presence of sheets (clusters) of Langerhans cells within the interstitium forming multiple tiny (less than 1 cm, sometimes less than 1 mm) ill-defined nodules with irregular edges (stellate nodules). Eosinophils can be numerous within the nodules. The occurrence of eosinophil-poor cases is one reason why "eosinophilic granuloma" was not a good term for this entity. Also, these lesions are NOT granulomas since they are collections of Langerhans cells, not epithelioid histiocytes. For the same reason, "Langerhans cell granulomatosis" is incorrect terminology. Langerhans cells are similar in some ways to ordinary macrophages (histiocytes), but their nuclei are more irregular and often have grooves, and resemble the kernel of a walnut. Cytoplasmic borders are somewhat poorly defined. Langerhans cells in lung lesions are often most numerous in the areas where the stroma looks slightly myxoid (like organizing pneumonia). In contrast, the more pink (collagenous/fibrous) the stroma becomes, the less likely it is that you will find Langerhans cells. Finally, in advanced lesions that are completely scarred, Langerhans cells are completely replaced by fibrous tissue, and cannot be demonstrated by immunohistochemistry. Other common pathologic findings in the background lung are pigmented macrophages within the alveolar lumens (airspaces). The terms "respiratory bronchiolitis" or a "DIP-like reaction" (a known misnomer) have been applied to these smoking-related pigmented macrophages. Other smoking-related changes that may be present in the background lung include emphysema and smoking-related interstitial fibrosis (SRIF). Vascular changes have also been described.
Immunohistochemical stains are not mandatory for the diagnosis but most pathologists do perform them to confirm that the cells in question are indeed Langerhans cells. Langerhans cells are positive for S-100, CD1a and Langerin (CD207). BRAF V600E mutations are only found in a subset of cases. In one study by Yousem and colleagues, 2/5 cases had a BRAF mutation. In another by Roden and colleagues, 7 of 25 (28%) cases were positive for a BRAF V600E mutation.
The diagnosis of pulmonary Langerhans cell histiocytosis is most commonly made by surgical lung biopsies, but the diagnosis can also be made by transbronchial lung biopsies. Diagnosis by core needle biopsies – as in this case – is uncommon, but has been previously reported (see references). The use of CD1a in BAL fluid is frequently cited as a way to make the diagnosis despite the fact that this approach is seldom if ever diagnostic in practice, and there is no evidence to support its use. This topic has been well reviewed in a recent article by Vehar and colleagues (see references).
The mainstay of treatment is smoking cessation. There are several possible outcomes, including complete or partial resolution of disease with smoking cessation, including radiologic resolution, which is well documented in the literature. Progressive scarring with lung function impairment occurs in a subset of patients. Secondary pulmonary hypertension may develop, and some patients develop smoking-related lung cancers. Other malignancies such as lymphoma have also been reported. Median survival was 12.5 years in a study of 102 adults.
Take home message for trainees: Consider the possibility of pulmonary Langerhans cell histiocytosis if you encounter a biopsy from a smoker with numerous small bilateral lung nodules.
References
Roden AC, Hu X, Kip S, et al. BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 2014;38:548-51.
Suri HS, Yi ES, Nowakowski GS, et al. Pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis 2012; PMID 22429393
Vehar S, Ribeiro Neto M, Culver D, et al. CD1a staining in bronchoalveolar lavage in pulmonary Langerhans cell histiocytosis: "barking up the wrong tree!" J Bronchol Interv Pulmonol 2022;29:e33-35.
Bonus open-access reference with links to references, notes on the history of the disease and numerous images: https://kikoxp.com/posts/20168/
Contributors
Director, Thoracic Pathology
Department of Pathology
Cleveland Clinic
Cleveland, OH, USA